Simcenter Culgi 2311 released! What’s new?
The design of chemical processes to produce novel materials or products comes with a bunch of critical engineering questions: What sequence of steps will be followed? What are the problematic phase interactions? What steps are needed to separate of purify products? What additives need to be added?
To answer these industry-scale process engineering questions with methods like process and system simulation, CAE methods like CFD, DEM, or FEM have become viable tools to predict the behavior of liquids, solids, and gases on the sub-process level. However, in times of increasing requirements on process efficiency, sustainability, and reliability those continuum scale simulations need to be more precise than ever. And so, CFD, DEM, and FEM simulations as well as system and process simulations need to be fed with accurate material properties and correct chemical interaction models to cater for competitive process engineering.
The challenge is that material properties and chemical interactions are buried on even smaller scales – in the underlying chemistry of molecules involved in those continuum dynamics and ultimately product manufacturing processes. Today, thanks to computational chemistry, materials scientists and chemical researchers can answer questions on the lab scale like: Which ingredient has what function? What intermediate step is necessary to reach the final product? Do the intermediate steps need additional ingredients? How will they change properties of the mix?
And so, for the ultimate design of industry-scale manufacturing processes, engineers must close the final gap between such computational chemistry simulations and the continuum and system scale methods. Designed as a seamless multiscale computational chemistry software solution Simcenter Culgi offers the tools to bridge that gap. With its latest release, Simcenter Culgi 2311 adds numerous new capabilities to design better materials and hence manufacturing processes and products, faster.
Learn how to use Support Center
New enhancements in Simcenter Culgi are aimed at helping you:

Model the complexity
Model any atom type with a Universal Force Field, be it organic, inorganic, or both
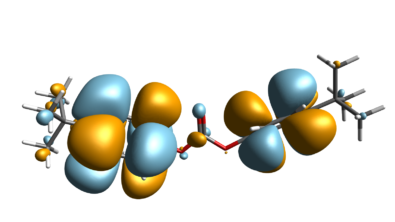
Atomistic scale Molecular Dynamics (MD) simulations require the definition of a force field to model the interaction between molecules. In traditional computational chemistry, users choose a force field model which is typically limited in its applicability to a given range of atoms or a family of molecules. It is up to the user to judge the best-suited force model from the literature in this approach. There is e.g. a commonly used potential/force field that works well for proteins, one for oil, etc. but there is no one-fits-all force field that avoids a priori preselection. This is critical when changing from organic to inorganic chemistry, as a specialized potential for one, no longer holds for the other and even more critical if the system at hand contains atoms from each family.
With the introduction of a Universal Force field in Simcenter Culgi 2311 we are overcoming all these challenges. A single, highly flexible force-field model now offers the ability to model any atom type – be it organic or inorganic or even a system containing both. This significantly increases the span of applications that can be modeled out of the box with no compromise on force field preselection. Batteries, glass, zeolites, catalysts are just a few examples of applications that will be enabled through such a universal force field description.
The new force field implementation has been validated against the common “traditional/specialized” force fields with respect to bond length, angles, etc. proving its general applicability with equal or even higher simulation fidelity as it allows for a more accurate parameterization.
Based on these capabilities you can leverage dedicated applications for screening battery materials, from polymers to lipid membranes. Even coarse grain simulations that are typically parametrized from MD simulations will benefit, increasing the out-of-the-box accuracy for applications such as battery slurry drying.
Explore the possibilities
Design and screen new molecules quickly, and easily with the Molecular generator
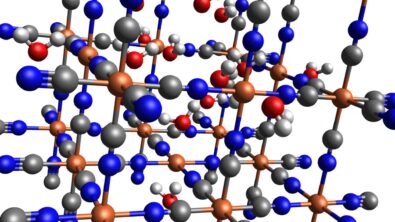
Quickly exploring and assessing new materials with reduced time-to-market is of paramount importance to stay competitive. While virtual screening with the help of computational chemistry is a game-changer compared to expensive experimental trial and error method, it still requires the virtual definition and rapid screening of a chemical design space to identify formulations that meet the target criteria.
With Simcenter Culgi 2311 you will get access to a novel chemistry exploration tool that allows you to perform such virtual screenings faster than ever. An intuitive drawing tool allows you to easily sketch even the most complex molecule concepts with functional group representatives using the Markush structure generator. By specifying the range of functional group parameters, you can generate a huge number of specific molecules from the {template structure} as potential candidates for a subsequent performance screening study or design exploration. The target properties (e.g. diffusivity, surface activity, etc.) can be defined as a goal for a given computational chemistry simulation, and- under the variation of the molecule – a design exploration tool like HEEDS will find the optimum molecular structure to meet the target.
Accessible to all user types and not limited to a specific industry the tool allows you to decrease formulation development time by 90% through a streamlined automated virtual screening of new chemistry and material solutions. Develop innovative formulations and optimize your intellectual property positioning by creating new materials and additives through virtual generation and testing.
Go faster
Leverage Dissipative Particle Dynamics (DPD) simulations’ potential with fully automatic parametrization
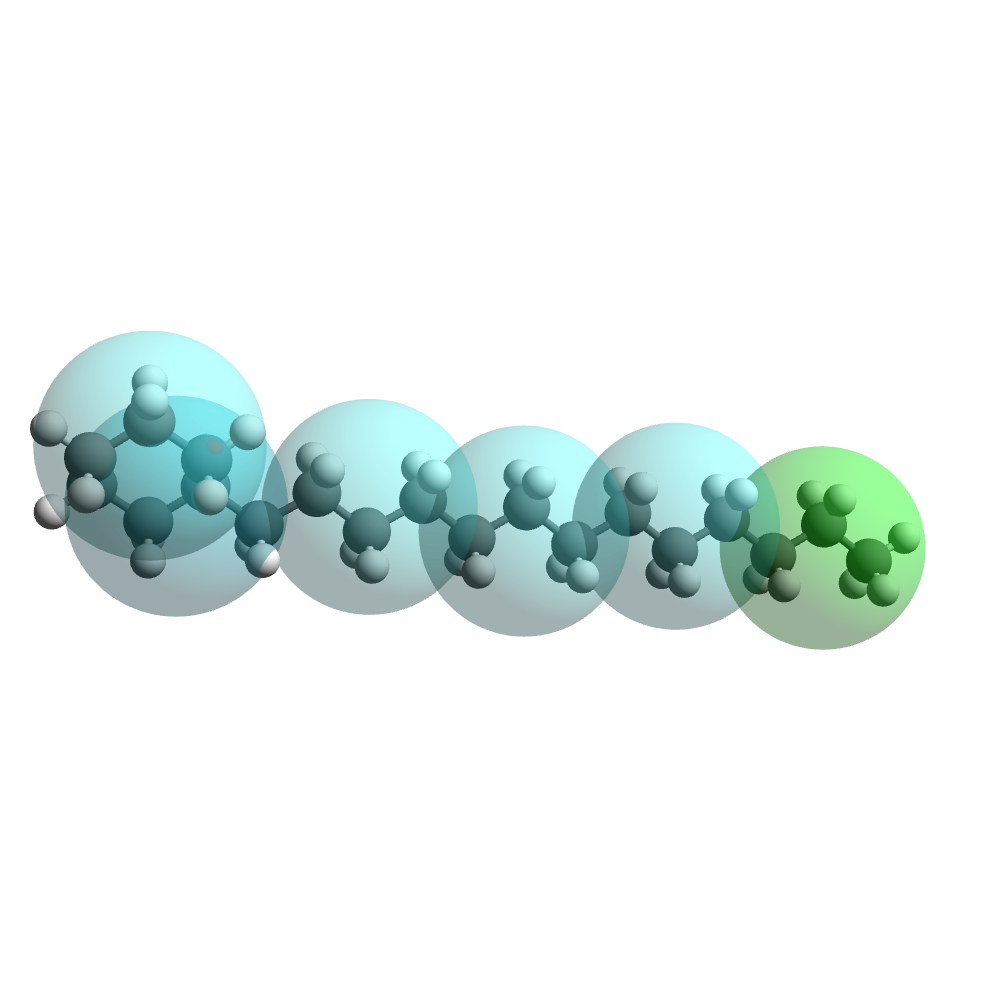
Across all industries and applications, for a fast assessment of chemicals on the mesoscale Dissipative Particle Dynamics (DPD) is the method of choice. However, the predictive value of these comparably efficient DPD simulations heavily relies on an accurate parametrization of the underlying inter- and intra-molecular interaction forces. While these interaction parameter sets can be determined from quantum calculations for electron distributions and atomistic scale molecular dynamics simulations, the manual determination of valid parameter sets – is the most painful and time-consuming part of such simulation workflows and requires deep expert knowledge. The rule of thumb is “1 day per molecule” for a skilled chemical engineer.
To overcome these challenges and leverage the full potential of DPD with ease, Simcenter Culgi 2311 comes with the fully automatic computation of interaction parameters from electron distributions. This brings down the preparation step of DPD parametrization from days to minutes for huge molecules and even down to seconds for smaller-sized molecules. The algorithm completely mitigates the need for trial-and-error tuning of hundreds of parameters. Its highly flexible designs allow for even the most complex and novel molecules to be automatically parametrized from electron distribution calculations. You can drive the complete Automatic parametrization from within the GUI of Simcenter Culgi and the complete parametrization will be written to a single parametrization file for subsequent DPD usage.
By taking the parameterization task away from the user, we simplify, accelerate, and automize the initialization process of any project. By this means not only will Simcenter Culgi 2311 let chemical engineers go significantly faster from quantum scale simulations to insightful meso-scale results to drive material property decisions, it will also enable a way broader range of engineers to leverage the power of such methodologies.
Improve computational efficiency through optimized code parallelization
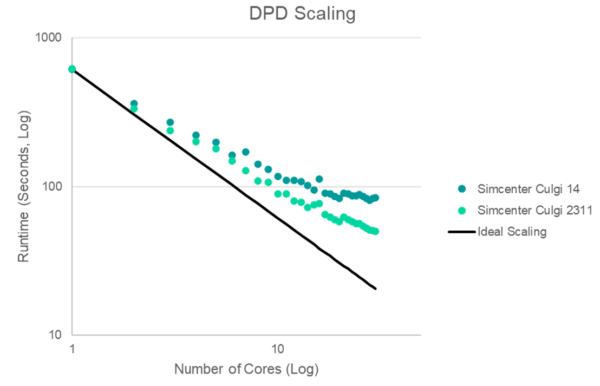
Chemistry simulations are becoming more complex and time-consuming due to the increasing fidelity of computational chemistry models. And so while a higher accuracy is highly desirable, increasing the throughput remains a constant need for computational chemistry simulations. Therefore the solvers in Simcenter Culgi have been designed for parallel computation and we are continuously seeking further speed-up opportunities through code optimization.
With Simcenter Culgi 2311 you will benefit from further parallelization optimization for atomistic and coarse-grained simulations to further accelerate formulation screening. Improving the calculation efficiency of force and energy of bonded interaction increases calculation speed of a DPD simulation by up to 30% for many applications with good scalability up to a high number of cores. Atomistic simulations can now be run up to 15% faster.
For any application, these acceleration methods increase your possibility to run more complex simulations and/or cover a longer period of time within a given turnaround timeframe.
These are just a few highlights in Simcenter Culgi 2311. Those features will enable you to design better products faster than ever, turning today’s engineering complexity into a competitive advantage.