Simcenter Culgi 15 released! What’s new?
Detailed insights into rheological processes, easy characterization of molecular vibrational infrared modes, high fidelity predictions of miscibility and permeability and accurate transport property predictions feeding CFD simulations. With the new release of Simcenter Culgi 15, computational chemists across industries can leverage a host of new features and enhancements to model the complexity of chemical systems while going faster in their simulations. The new release allows computational chemists and data scientists to explore the possibilities of any given chemical system to come up with novel materials and tailored material properties.
New enhancements in Simcenter Culgi are aimed at helping you:
Learn how to use Support Center

Explore the possibilities
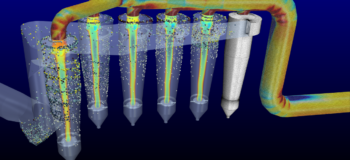
Understand flow patterns in particle-based simulations
Understanding the mixing behavior as a function of shear, including aggregation effects, reaction dynamics and crystallization effects is crucial in the development of formulations, polymer blends and emulsions. Computational chemists across industries, including Food & Beverage, Chemical, Personal Care, Polymer, Pharmaceutical, Oil & Gas, need to deliver detailed insights into such rheological processes to ensure optimum product performance.
However, the nature of the underlying transient molecular dynamics often makes it difficult to comprehend the flow patterns that are driven by the stochastic motion of individual molecules or atoms. But, gathering insights into those large-scale motion patterns is crucial to understanding rheological processes and properties.
Simcenter Culgi 15 now provides the ability to calculate time- and space-interval averaged scalar (e.g., density, X-component of velocity) and vector (e.g., velocity, force) fields of selected atoms or beads from a particle-based system simulation. With this new particle motion average, computational chemists can now comprehend rheology results faster, identify patterns and easily visualize molecular motion patterns in a formulation.
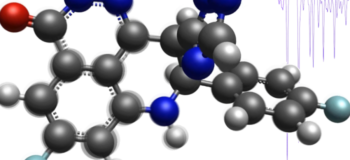
Easily characterize materials through Vibrational Infrared (IR) mode analysis
A major task of computational chemists in Food & Beverage, Chemical or Polymer segment is to characterize highly complex materials and correlate their molecular characteristics to the final material or product performance. Applications like polymer curing, reactive blends, formulations and adhesives heavily depend on a detailed upfront characterization of the molecular properties to ensure their functionality in the end product.
One method of significant importance for such material characterization efforts is Vibrational Infrared (IR) analysis. Simcenter Culgi 15 offers computational chemists and lab scientists a fast and easy molecular vibrational analysis for even the most complex molecular structures. The tool is providing information about normal modes of the molecular vibrations and the corresponding infrared (IR) spectrum for fundamental frequencies based on the harmonic oscillator model. In addition to quantitative analysis, computational chemists can quickly gain and communicate detailed three-dimensional insights through the embedded animation of the normal modes of vibration.
Model the complexity
Predict miscibility for any chemical formulation using the Cohesive Energy Density method
Understanding the miscibility of liquids is critical in many industries, whose business models heavily depend on the mixing, handling, and transport of those multi-component liquids. This includes Food & Beverage, Chemical, Personal Care, Polymer, Pharmaceutical and the Oil & Gas industries. Not being able to provide reliable, yet fast predictions of liquid miscibilities puts companies at risk to face significant inefficiencies or even a complete failure of their product.
Hence having access to a quick and accurate solubility calculator for any given chemistry has a massive direct business impact to those industries. While traditional methods, including statistical analysis and look-up table-based approaches (e.g. group contribution methods or UNIFAC) are a quick way for established chemistry they cannot be used for novel and more sophisticated chemical systems.
Simcenter Culgi 15 now complements the traditional group contribution method with the Cohesive Energy Density method. This virtual screening tool helps R&D scientists to predict solubility parameter of liquids by smartly extracting inter-molecular forces from Molecular Dynamics simulations for any given chemistry.
Ref: Hildebrand and Hansen solubility parameters from Molecular Dynamics with applications to electronic nose polymer sensors,
M. Belmares, M. Blanco,W. A. Goddard III, R. B. Ross, G. Caldwell, S.-H. Chou, J. Pham, P. M. Olofson, Cristina Thomas, J Comput Chem 25: 1814–1826, 2004.
Better permeability predictions using the Nosé-Hoover chain (NHC) algorithm
Accurate predictions of diffusion are required to understand the permeability of common gas-phase molecules and food simulants through polymer matrices. Understanding and quantifying barrier properties of materials is critical in many applications in the food, beverage, personal care and cosmetics industries.
Simcenter Culgi 15 now offers a novel method to run molecular dynamics simulation at constant number of particles, constant volume and constant temperature (NVT). Computational chemists running NVT can now leverage the Nosé-Hoover chain (NHC) algorithm replacing the Nosé-Hoover algorithm. NHC ensures superior numerical stability while predicting a correct canonical distribution giving an accurate diffusion and viscosity leading to better material choices.

Stay integrated
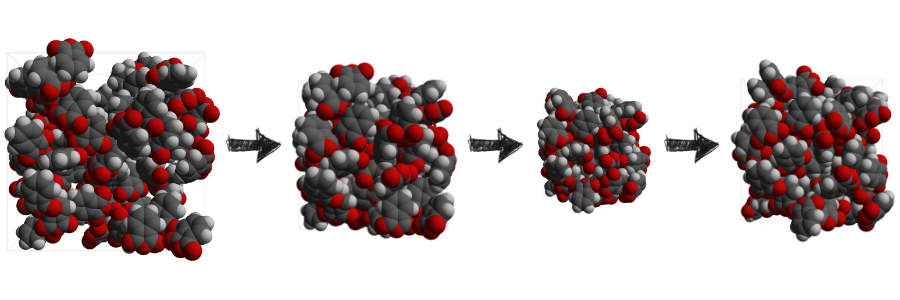
Predict critical transport properties for CFD simulations
Transport properties such as viscosity are a critical input quantity to computational fluid dynamics simulations. The accurate computation of those properties through particle dynamics simulations offers huge potential to replace the often difficult, costly, and time-consuming experimental assessment. Furthermore, such simulations mitigate the error-prone extrapolation of such properties to experimentally inaccessible temperature and pressure ranges.
With Simcenter Culgi 15 a new method allows computational chemists to control the thermostat of a dissipative particle dynamics (DPD) coarse-grained simulation. By offering the ability to enable/disable the projection of the friction and random forces onto the line joining interacting beads in the pair-wise DPD thermostat simulation chemists have more control over the computation of transport properties such as the viscosity. This, together with the ability to lump together atomic groups into beads, allows coarse-grained simulations to reach speed-ups of three orders of magnitude. The method enables chemists to simulate much larger length and time scales while maintaining sufficient accuracy and stability. The resulting fast and accurate prediction of transport properties at the coarse-grained level can then be leveraged for increased accuracy in CFD simulations using Simcenter STAR-CCM+. Those simulations bridge different departments in a company and help them understand their respective results.