MDR and Intelligent Design Control
Are You Ready?
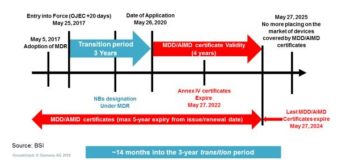
I recently attended a BSI roadshow event in London where the FDA, BSI, and British MDRA organizations made an attempt to educate me on the implication of the Medical Device Regulation (MDR). The following is their summary of the timeline for activities over the next several years. The Invitro Diagnostic Regulation (IVDR) is similar with a longer transition and shorter certificate validity ending on May 7, 2024 like the medical device timeline.
 Figure 1 – MDR Implementation Timeline – BSI Medical Device Roadshow
Figure 1 – MDR Implementation Timeline – BSI Medical Device Roadshow
We are currently about a year into the transition period, so we are well on our way to a significant regulatory hard stop. The regulation represents the largest update in international regulatory change in almost 25 years, so I thought it might be helpful to discuss some of the major changes, and how tools can be utilized to minimize the pain of transition.
“Significant Changes”
The MDR is essentially silent on the triggers for conducting a new clinical investigation when a device is changed or modified. The language specifies a requirement for clinical investigation when a “significant change” is made. Likely factors would be related to Article 62 section 1.
Suitability of its intended use and achievement of the intended performance; to verify that the clinical benefits as specified have not been altered; that the device is clinically safe; the undesirable side effects continue to constitute acceptable risks when weighed against the benefits of the device.
This calls into question the extent to which a device that is qualified under the MDD standard during the 4 year certificate validity period would need to be re-qualified with MDR clinical requirements if “significant changes” were made. In any case a detailed regulatory impact analysis should be completed for all of your devices to clearly understand the requirements for qualification under the MDR standard. At a minimum it would provide the plan required to quickly act on re-qualification as the need arises. Needless to say, this is an extremely detailed piece of work, and Siemens could help you to provide systems around the product design control, and regulatory impact analysis.
Post-Market Surveillance (PMS) and Risk
The PMS section of the new regulation is new and comprehensive. PMS has been elevated to a structural focus of the regulation.
Manufacturers shall report, by means of the electronic system referred to in Article 92, any statistically significant increase in the frequency or severity of incidents that are not serious incidents.
Draw your attention to two points of this requirement. The first is “any statistically significant increase.” In isolation this is not new. However, when combined with “incidents that are not serious incidents” we find that statistical analysis is no longer limited to serious incidents. In the past we could focus on relatively few serious issues. Now we have a tipping point. The requirement is to do a statistical analysis on ALL of the incidents. For this serious consideration should be given to automated systems to provide exception management on the entire design stack.
PMS Plan – Article 84 – Annex III Section 1.1
- Information concerning the evaluation of serious incidents, including information from Periodic Safety Update Report (PSURs), and field safety corrective actions;
- Records referring to non-serious incidents and data on any undesirable side-effects;
- Information from trend reporting;
- Relevant specialist or technical literature, databases and/or registers;
- Information, including feedbacks and complaints, provided by users, distributors and importers; and
- Publicly available information about similar medical devices
PMS Report – Article 85
- Surveillance data; rationale and description of CAPA actions taken
- Periodic Safety Update Report (PSUR)
- Evaluation of the description of the intended purpose of the device
- Evaluation of the device’s benefits to the patient
- Quantification of benefit(s) to the patients
- Probability of the patient experiencing one or more benefit(s)
- Duration of effects(s)
- Evaluation of the clinical risks of devices (extent of risk(s) / harm(s), the following should be addressed individually and in aggregate):
- Severity, number and rates of harmful events
- Probability of harmful events
- Duration of harmful events
- Risk from false-positive or false-negative results (diagnostic medical devices)
- Evaluation of acceptability of the benefit/risk profile
These systems need to be linked to provide for a functionally relevant, predictive performance evaluation. As you might imagine a regulatory change of this magnitude includes much more than I can cover in the context of a blog, but hopefully this provides you with a few of the weightier issues of concern to medical device manufacturer systems development work. If you are interested in mature systems that provide solutions to these difficult problems take a look at the following link:


